
The final date for all MDD-certified devices to be certified under the new MDR is May 25, 2024. Once a medical device is EU MDR compliant (even prior to this date), it can be certified under the MDR. However, it is not mandatory to obtain EU MDR certification, if the MDD certificate for the device is still valid until May 25, 2024. If the EU MDD certificate expires before 25 May 2024, then such a medical device needs to be recertified according to the EU MDR. Hence, the last date to obtain EU MDR certification is May 25, 2024. After this date, all devices on the market must be certified under the EU MDR.
Key Changes
After almost 25 years, the EU MDD was fundamentally revised. This was essential to establishing a more transparent, robust, sustainable, and predictable regulatory framework for improving the overall level of health and safety of medical devices, without compromising on innovation in the industry. The new EU MDR legislation imposes regulatory obligations unlike EU MDD (which was purely directive in nature). Some of the key changes imposed by the EU MDR are:
Product Classification
-
-
- EU MDR specifies new rules for substance-based devices. Devices with substances to be absorbed by the body are under the new classification system. This has caused many devices to be reclassified into higher-risk classes.
- The product classification is expanded with new as well as revised terms and rules. Therefore, you should reassess the current classification route of your medical device. It may now fall under a different rule, and the Class of the medical device might be affected. Moreover, there are new classifications have been added such as Class 1m (m stands for a Class 1 device with measuring function), Class 1s (s stands for a Class 1 device with sterile function), there is a new Class 1r (r stands for a Class 1 reusable surgical instruments). In addition, stand-alone software falls under Class IIa or higher according to the MDR, which was classified as Class I according to the MDD.
- EU MDR specifies new rules for substance-based devices. Devices with substances to be absorbed by the body are under the new classification system. This has caused many devices to be reclassified into higher-risk classes.
-
Clinical Evaluation Process
-
-
- Manufacturers should conduct a clinical evaluation process in accordance with the requirements stated in Article 61 and Annex XIV, including a PMCF.
- Tighter areas of clinical investigations and post-marketing follow-ups.
- Clinical evidence needs to be up-to-date, clear, convincing, and publicly available.
- For Class III devices and implantable devices, the post-market clinical follow-up evaluation report, the summary of safety and clinical performance shall be updated at least annually.
- The requirements for clinical evaluations and trials are expanded significantly in the EU MDR. If a medical device is a high-risk product, clinical data should be collected and evaluated using the medical device, if competitor device data is used for clinical evaluation activities, it should be justified including why and how they are similar.
- Manufacturers should conduct a clinical evaluation process in accordance with the requirements stated in Article 61 and Annex XIV, including a PMCF.
-
PMS (Post-Market Surveillance) System
-
-
- Manufacturers should implement a post-market surveillance system with a continuous evaluation and improvement loop that links to continuous reviews of risk management and an annual update of a public summary of safety and performance, as well as clinical evaluation. This is a standard part of EN ISO 14971 but is now mandatory for all according to Annex III.
- The MDR focuses on the area of post-market surveillance (i.e., PMS) as well as the general reporting obligations (i.e., Vigilance). The PMS plan and corresponding report are required for Class I devices whereas a PSUR (periodic safety update report) is also required for Classes IIa, IIb, and III.
- Manufacturers should implement a post-market surveillance system with a continuous evaluation and improvement loop that links to continuous reviews of risk management and an annual update of a public summary of safety and performance, as well as clinical evaluation. This is a standard part of EN ISO 14971 but is now mandatory for all according to Annex III.
-
Quality management system
-
-
- Manufacturers of devices, other than investigational devices, shall establish, document, implement, maintain, keep up to date, and continually improve a quality management system that shall ensure compliance with this regulation in the most effective manner and in a manner that is proportionate to the risk class and the type of device.
-
Person Responsible for Regulatory Compliance
-
-
- Manufacturers should have at least one responsible person available within the organization who has the requisite expertise in the medical devices industry.
- The PRRC (person responsible for regulatory compliance) is responsible for the EU MDR compliance of their organization. PRRC ensures that the technical
documentation and EU DoC (declaration of conformity) are prepared and updated regularly, and post-market surveillance activities are documented (see Article 15).
- Manufacturers should have at least one responsible person available within the organization who has the requisite expertise in the medical devices industry.
-
Notified Bodies
-
-
- These are organizations designated by EU member states. A notified body performs the assessment of product compliance with all the obligatory standards and procedures.
- The major change introduced by MDR and IVDR relates to the supervision of notified bodies, because of this change, a substantial number of the current notified bodies may potentially lose their designation or see changes in the scope of their authority.
- Notified bodies will have additional responsibilities as they will have to consult with the European Commission on clinical evaluation and conduct surveillance assessments.
- Notified bodies will continue to provide assessment only of higher-class medical devices.
- Notified bodies are liable in case of any issue that they missed on your PMS system. So that they tend to spend more time reviewing your PMS system and PMS data.
- These are organizations designated by EU member states. A notified body performs the assessment of product compliance with all the obligatory standards and procedures.
-
Economic Operators
-
-
- New regulations provide guidelines for all stakeholders on how to execute assigned duties and help organize responsibilities among suppliers, importers, subcontractors, assemblers, and EU-authorized representatives.
- This is the most welcomed change by all parties as it makes processes like technical documentation, complaint submissions, labeling, and post-market surveillance much clearer. These changes are covered in Articles 10, 11, 13, 14, and 30.
- New regulations provide guidelines for all stakeholders on how to execute assigned duties and help organize responsibilities among suppliers, importers, subcontractors, assemblers, and EU-authorized representatives.
-
EUDAMED Changes
-
-
- EUDAMED is the European Union’s database for medical devices, which was founded in 2011. Please note that EUDAMED is not a public database.
- It is a web-based platform that stores all relevant regulatory information for medical devices. The regulatory information is received from manufacturers and notified bodies. The main reason behind the establishment of EUDAMED is to facilitate information exchange between the European Commission and competent authorities in the EU states.
- The information includes the registry of manufacturers, declaration of conformity, authorized representatives, and incidents or near-incidents reported during the use.
- The EUDAMED database will now also store information related to post-market surveillance activities, periodic safety update reports, safety, and clinical performance studies. It will also provide more detailed information on manufacturers, clinical investigation data, and device registrations. Article 33 covers details on changes related to the EUDAMED database.
- EUDAMED is the European Union’s database for medical devices, which was founded in 2011. Please note that EUDAMED is not a public database.
-
UDI (Unique Device Identification) System
-
-
- The implementation of UDI (Unique Device Identification) into EU MDR has been long-awaited and is widely considered essential.
- The addition of a UDI system intends to increase the traceability of all medical devices available in the market by placing a unique code on the device label.
- Each medical device should have a UDI-DI (UDI-Device Identifier) and UDI-PI (UDI-Production Identifier). Furthermore, each UDI should be submitted to the UDI database.
- UDI offers several benefits and will be primarily utilized for reporting serious safety incidents and identifying counterfeit medical devices.
- The details of this new system are covered in Articles 18, 19, 27, and 87.
- The implementation of UDI (Unique Device Identification) into EU MDR has been long-awaited and is widely considered essential.
-
In short, the requirements of MDR are as follows:
- Annex I specifies the safety and performance requirements.
- Annex II specifies the requirements of certain technical documentation.
- The manufacturer should choose between these four approaches:
-
-
- Annex IX: a conformity assessment (by a notified body) will have to be completed on the QMS against ISO 13485 and the technical documentation. Then, if the MD is implantable, medicinal, human origin, animal origin, or absorbed/dispersed, section 5 of Annex IX applies, and there are extra special procedures.
- Annex IX: Same as above, but it is not implantable, medicinal, human origin, animal origin, or absorbed/dispersed: No need for the extra special procedures.
- Annex X, production is audited, and a representative sample of production is tested by the notified body. And, as described in Annex XI, Part A: A conformity assessment (by a notified body) will have to be completed on the production quality system (excluding the design & verification activities).
- Annex X, production is audited, and a representative sample of production is tested by the notified body. And, as described in Annex XI, Part B: A verification (by a notified body) will have to be completed on product conformity (based on 100% testing), for example on a small production batch.
- Annex IX: a conformity assessment (by a notified body) will have to be completed on the QMS against ISO 13485 and the technical documentation. Then, if the MD is implantable, medicinal, human origin, animal origin, or absorbed/dispersed, section 5 of Annex IX applies, and there are extra special procedures.
-
- Annex IV – This annex specifies the requirements for the declaration of conformity.
- Annex V – This annex specifies the requirements for the CE mark.
EUDAMED
The European database on medical devices – short EUDAMED – is one of the key elements of the MDR and IVDR. EUDAMED aims to enhance transparency overall, including better access to information for the public and healthcare professionals. It further intends to enhance coordination between the various states in the EU.
The EUDAMED registration modules became active in 2021, and it is important to note that the process adds significant work for most manufacturers. You should start your registration process earlier, as it is not only a registration process and the EUDAMED modules should be aligned with your internal systems. Currently, there are three live modules out of the six total planned modules:
- Actor registration (live)
- UDI (unique device identification) and device registration (live)
- Notified bodies and certificates (live)
- Clinical investigations and performance studies
- Vigilance and market surveillance
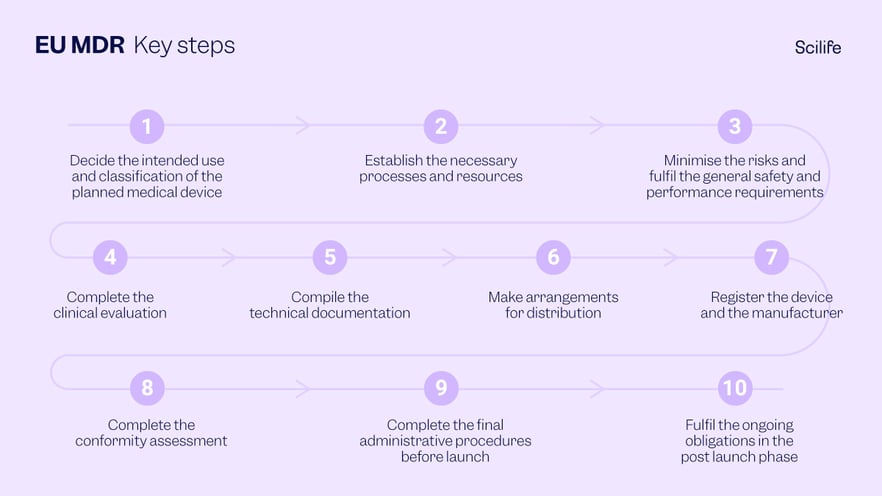
Important Steps
The official website for EU MDR lays out the following ten important steps to comply with the regulation.
Conclusion
As we are monitoring the legislative developments closely, our aim is to help our life sciences and medical device companies achieve total EU MDR compliance through the Scilife platform.
We hope that our clients will take full advantage of our services, as EU MDR also encourages the manufacturers to leverage support from their suppliers.
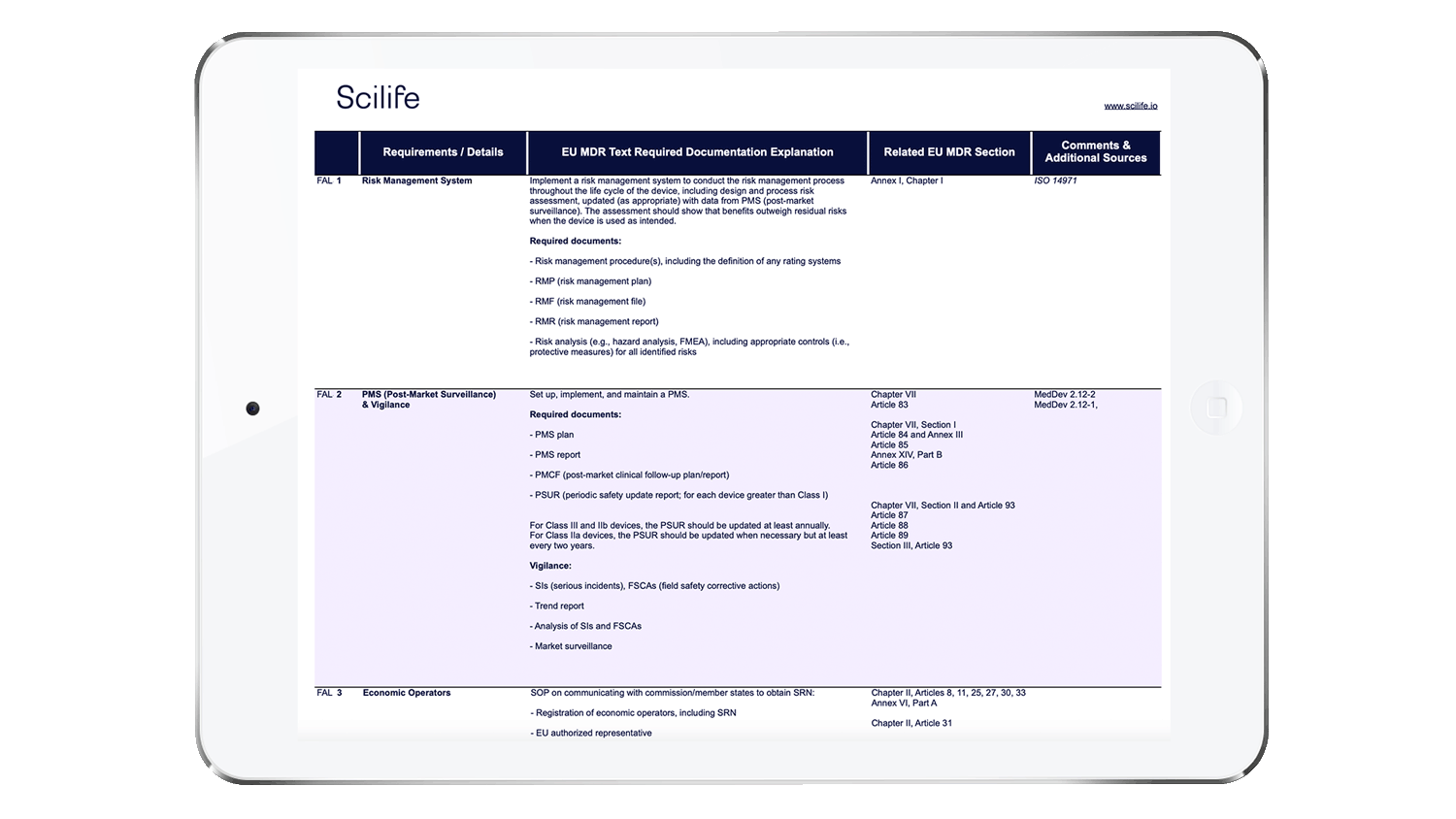
We have prepared a checklist to help you self-assess your readiness for EU MDR key changes.
|
|

A well-implemented QMS in your organization can save you hours when creating and organizing the documentation required by authorities and the EU MDR.
With each new regulation, more effort is needed from manufacturers, especially from their QA and RA professionals. You should determine current and future needs and decide to implement a QMS that allows you to manage day-to-day tasks along with planned or unexpected requirements that might take weeks or months to comply with.
To get ready for the MDR audit, we recommend you start by analyzing your current QMS. If your QMS does not pass or if you have any uncertainties, Scilife is an ideal option for life sciences and medical device companies. We offer a dedicated platform that goes above and beyond an advanced QMS by improving all your processes as well as your quality management.
Ready to get fully EU MDR compliant with Scilife Smart QMS for Medical Devices?




